傾儖僣僴僀儅乕昦偺暘巕昦懺
| 傾儖僣僴僀儅乕昦偺暘巕昦懺偵娭偟偰偼尰嵼傑偱傾儈儘僀僪(A兝)僇僗働乕僪壖愢偑桳椡偱偡丅偡側傢偪丄A兝偺擼撪夁忚拁愊偑暘巕昦懺偺崻尮偱偁傞偲偝傟偰偄傑偡丅杮尋媶幒偱偼庡偵
恄宱嵶朎偺A兝惗惉偵懳偡傞暘巕惂屼儊僇僯僘儉偺夝柧偲偦傟偵婎偯偄偨帯椕朄奐敪偵徟揰傪摉偰偨僾儘僕僃僋僩傪恑傔偰偄傑偡丅
|
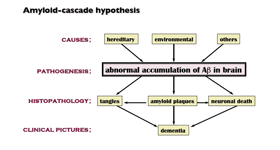 |
乮I乯恄宱嵶朎偺A兝惗惉傪惂屼偡傞暘巕儊僇僯僘儉偺夝柧
| 擼偺A兝偼庡偵恄宱嵶朎偵傛偭偰嶌傜傟傑偡丅A兝偼慜嬱懱僞儞僷僋幙偱偁傞APP偑兝僙僋儗僞乕僛偲兞僙僋儗僞乕僛偵傛傝愗抐偝傟偰偱偒傞40側偄偟42傾儈僲巁偐傜惉傞億儕儁僾僠僪偱偡丅
偙偺偆偪丄兞僙僋儗僞乕僛偼APP偺枌娧捠椞堟傪壛悈暘夝偡傞偲偄偆戝偒側摿挜傪傕偭偰偄傑偡丅枌撪偲偄偆慳悈惈娐嫬偵偍偄偰僞儞僷僋幙傪壛悈暘夝偡傞僾儘僥傾乕僛偑偁傞偙偲偼嬤擭偵側偭偰柧傜偐偵側偭偰偒偨怴偟偄抦尒偱偡丅偙偺傛偆側僾儘
僥傾乕僛偵傛傞愗抐偑挷愡偝傟傞儊僇僯僘儉傪偼偠傔丄嵶朎偵杮棃旛傢偭偰偄傞A兝惗惉惂屼偺暘巕婡峔傪抦傞偙偲偐傜丄暃嶌梡偺側偄A兝惗惉梷惂朄偺奐敪傪栚巜偟傑偡丅 |
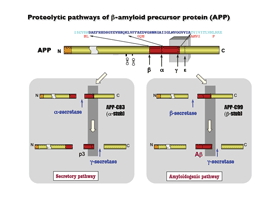 |
乮II乯兞僙僋儗僞乕僛偺愗抐晹埵摿堎惈傪惂屼偡傞嵶朎撪場惈儊僇僯僘儉偺夝柧
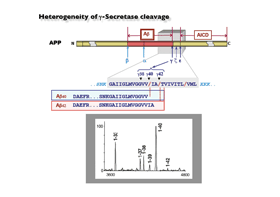
| A兝偵偼兞僙僋儗僞乕僛愗抐晹埵偺偢傟偵傛傝傾儈僲巁嵔挿偺堎側傞暘巕庬偑抦傜傟丄栺90%傪愯傔傞偲A兝40偲栺10%偵偁偨傞A兝42側偳偑専弌偝傟偰偄傑偡丅傾儖僣
僴僀儅乕昦偺昦懺偵偼擼撪A兝検偲偲傕偵A兝42/ A兝40斾偑廳梫偱偁傞偙偲偑敾柧偟偰偒傑偟偨丅偡側傢偪丄昦懺傪庝婲偡傞偺偼A兝42偱偁傝丄A兝40偼傓偟傠昦懺偺恑峴傪梷惂偡傞偲偝傟傑偡丅廬偭偰丄
兞僙僋儗僞乕僛偺愗抐晹埵摿堎惈傪寛傔傞儊僇僯僘儉傪柧傜偐偵偟丄偙傟傪恖堊揑偵僐儞僩儘乕儖偡傞偙偲偑偱偒傟偽棟憐揑側帯椕朄奐敪偵偮側偑傞壜擻惈偑偁傝傑偡丅恄宱嵶朎偑杮棃傕偮惂屼儊僇僯僘儉偺夝柧偐傜帯椕朄偺奐敪傪栚巜偟傑偡丅 |
 |
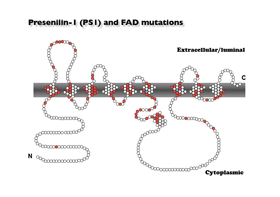
乮III乯僾儗僙僯儕儞偺婡擻夝愅
| 壠懓惈傾儖僣僴僀儅乕昦偺尨場堚揱巕偱偁傞僾儗僙僯儕儞(Presenilin-1 and Presenilin-2)偼丄枌僞儞僷僋幙傪僐乕僪偟丄懡偔偺昦尨曄堎偑抦傜傟偰偄傑偡乮塃恾嶲徠丅愒娵偑昦尨曄堎偺
抦傜傟傞巆婎乯丅婡擻偲偟偰偼兞僙僋儗僞乕僛暋崌懱偺峔惉僞儞僷僋幙偲偟偰怗攠晹埵傪採嫙偡傞偙偲偑抦傜傟傞偲摨帪偵丄懡婡擻僞儞僷僋幙偲偟偰丄傾億僩-僔僗傗嵶朎撪Ca峆忢惈偺堐帩丄枌僞儞僷僋幙桝憲側偳偵娭梌偡傞偲偝傟偰偄傑偡丅懡偔偺
婡擻傪偳偺傛偆偵巊偄暘偗傞偺偐丄懡婡擻傪傕偮偙偲偺惗暔妛揑側堄媊偼壗偐側偳嫽枴偁傞栤戣偱偡丅 |
 |
乮IV乯屒敪惈傾儖僣僴僀儅乕昦偺昦場偵娭傢傞暘巕偺摨掕
| 傾儈儘僀僪(A兝)僇僗働乕僪壖愢偵偍偄偰丄A兝拁愊偺忋棳偵埵抲偡傞崻杮尨場偵偮偄偰偼堚揱巕偺曄堎傗懡宆側偳愭揤揑側梫場偑抦傜傟傞埲奜偼傎偲傫偳夝柧偝傟偰偄傑偣傫丅傾儖僣僴僀儅乕昦徢椺偺
偆偪戝敿偼堚揱揑梫場偑悇應偱偒側偄屒敪惈偺徢椺偱偁傞偙偲偐傜丄屻揤揑梫場偺夝柧偼椪彴堛妛揑偵廳梫側壽戣偱偡丅偮傑傝丄偦偺夝柧偼懡偔偺徢椺偺敪徢傪梊杊偱偒傞僸儞僩偵傕側傝傑偡丅 |
乮V乯榁壔偲恄宱曄惈偺儕僗僋
| 堚揱巕偵柧傜偐側昦尨曄堎偑偁傞応崌偱傕丄懡偔偼拞崅擭偵側偭偰傾儖僣僴僀儅乕昦傪敪徢偟傑偡丅愭揤揑曄堎偵傛傞昦懺偑椪彴徢忬傪弌偡偵摓傞傑偱偵偦傟偩偗偺婜娫傪梫偡傞偺偐丄偁傞偄偼榁壔偲
偄偆場巕偑壛傢傞偙偲偑敪徢偵寛掕揑偵娭傢傞偺偐偼晄柧偱偡丅尰嵼丄惗暔妛揑側榁壔尰徾偑暘巕儗儀儖偱夝柧偝傟巒傔偰偄傞攚宨偐傜丄偙偺揰傪柧傜偐偵偱偒傞壜擻惈偑偱偰偒傑偟偨丅 |